publications
publications by categories in reversed chronological order. generated by jekyll-scholar.
2025
-
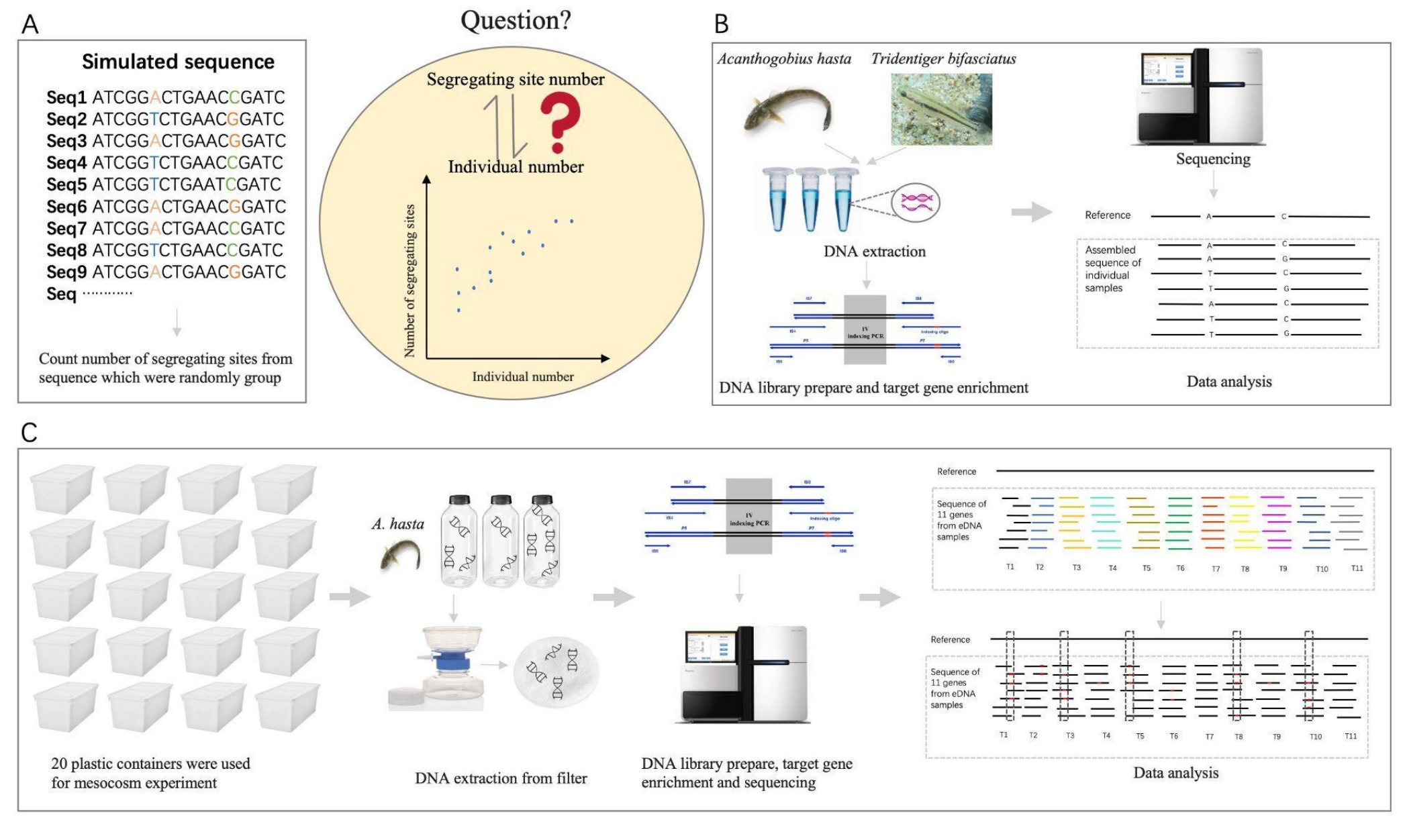 Estimation of species abundance based on the number of segregating sites using environmental DNA (eDNA)Qiaoyun Ai, Hao Yuan, Ying Wang, and 1 more authorMolecular Ecology Resources, 2025
Estimation of species abundance based on the number of segregating sites using environmental DNA (eDNA)Qiaoyun Ai, Hao Yuan, Ying Wang, and 1 more authorMolecular Ecology Resources, 2025The advance of environmental DNA (eDNA) has enabled rapid and non-invasive species detection in aquatic environments. Although most studies focus on species detections, some recent studies explored the potential of using eDNA concentration to quantify species abundance. However, the differential individual DNA contribution to eDNA samples could easily obscure the concentration-species abundance relationship. We propose using the number of segregating sites as a proxy for estimating species abundance. Since segregating sites reflects the genetic diversity of the population, which is less sensitive to differential individual DNA contribution than eDNA concentration. We examined the relationship between the number of segregating sites and species abundance in silico, in vitro, and in situ using two brackish goby species, Acanthogobius hasta and Tridentiger bifasciatus. Analyses of the simulated data and in vitro data with DNA mixed from a known number of individuals showed a strong correlation between the number of segregating sites and species abundance (R2 > 0.9; P < 0.01). Results from the in situ experiment further validated the correlation (R2 = 0.70, P < 0.01), and such correlation was not affected by biotic factors, including body size and feeding behavior (P > 0.05). Results of the cross-validation test also showed that the number of segregating sites predicted species abundance with less bias and variability than the eDNA concentration. Overall, the number of segregating sites correlates stronger with species abundance and also provides a better estimate than eDNA concentration. This advancement can significantly enhance the quantitative capabilities of eDNA technology.

2024
-
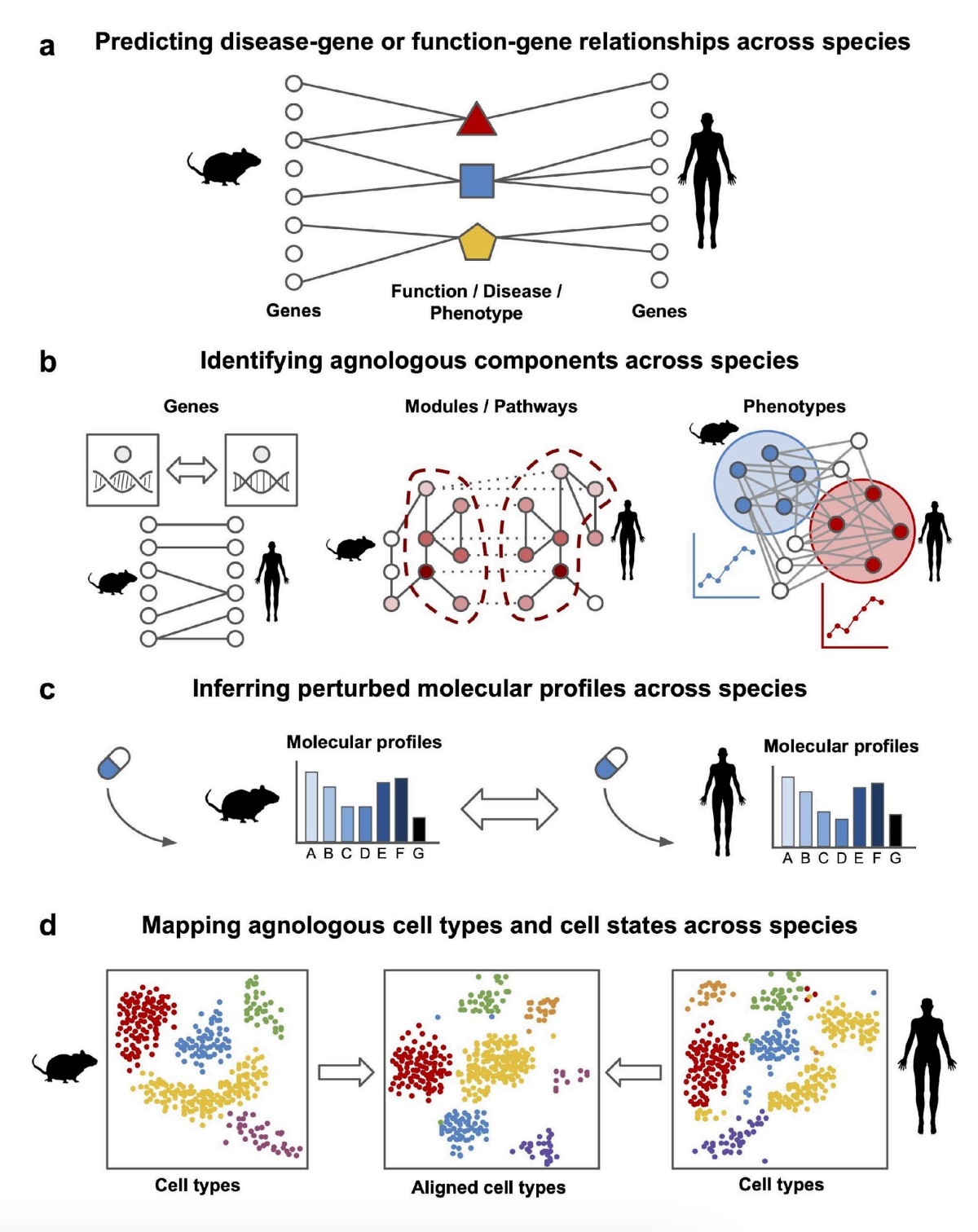 Computational strategies for cross-species knowledge transfer and translational biomedicineHao Yuan, Christopher A Mancuso, Kayla Johnson, and 2 more authorsarXiv preprint arXiv:2408.08503, 2024
Computational strategies for cross-species knowledge transfer and translational biomedicineHao Yuan, Christopher A Mancuso, Kayla Johnson, and 2 more authorsarXiv preprint arXiv:2408.08503, 2024Research organisms provide invaluable insights into human biology and diseases, serving as essential tools for functional experiments, disease modeling, and drug testing. However, evolutionary divergence between humans and research organisms hinders effective knowledge transfer across species. Here, we review state-of-the-art methods for computationally transferring knowledge across species, primarily focusing on methods that utilize transcriptome data and/or molecular networks. We introduce the term "agnology" to describe the functional equivalence of molecular components regardless of evolutionary origin, as this concept is becoming pervasive in integrative data-driven models where the role of evolutionary origin can become unclear. Our review addresses four key areas of information and knowledge transfer across species: (1) transferring disease and gene annotation knowledge, (2) identifying agnologous molecular components, (3) inferring equivalent perturbed genes or gene sets, and (4) identifying agnologous cell types. We conclude with an outlook on future directions and several key challenges that remain in cross-species knowledge transfer.
-
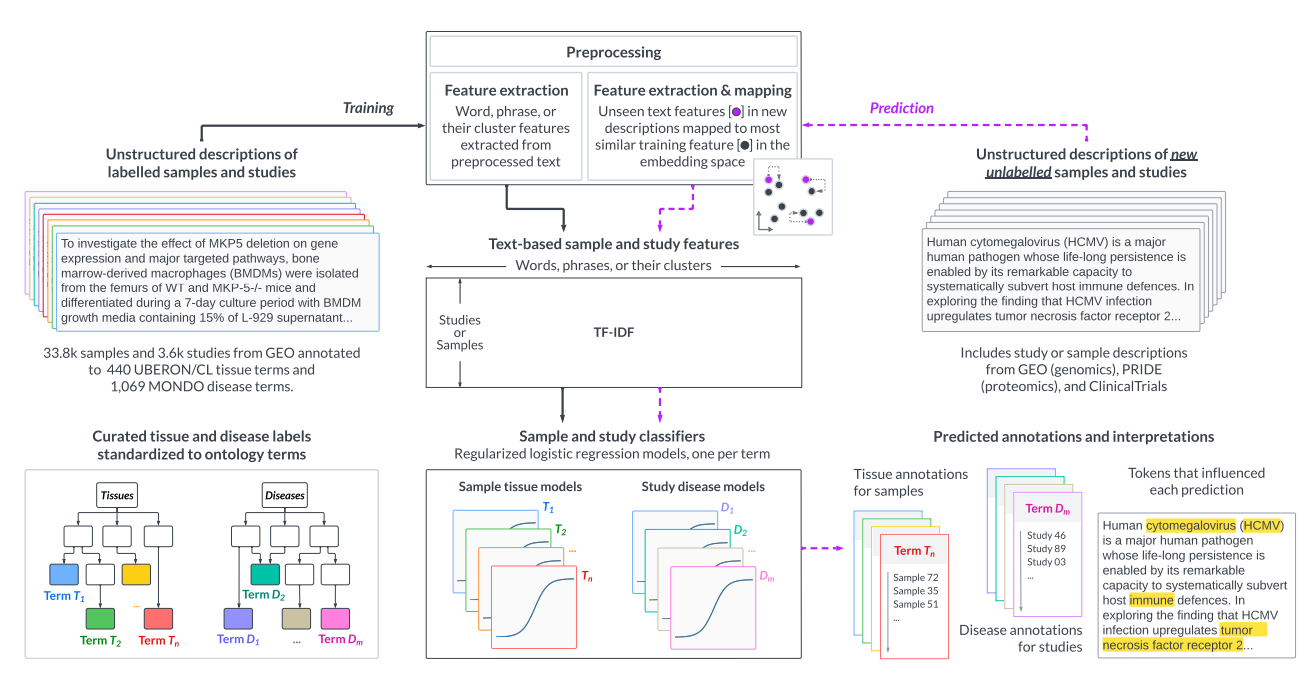 Annotating publicly-available samples and studies using interpretable modeling of unstructured metadataHao Yuan, Parker Hicks, Mansooreh Ahmadian, and 3 more authorsBriefings in Bioinformatics, 2024
Annotating publicly-available samples and studies using interpretable modeling of unstructured metadataHao Yuan, Parker Hicks, Mansooreh Ahmadian, and 3 more authorsBriefings in Bioinformatics, 2024Reusing massive collections of publicly available biomedical data can significantly impact knowledge discovery. However, these public samples and studies are typically described using unstructured plain text, hindering the findability and further reuse of the data. To combat this problem, we propose txt2onto 2.0, a general-purpose method based on natural language processing and machine learning for annotating biomedical unstructured metadata to controlled vocabularies of diseases and tissues. Compared to the previous version (txt2onto 1.0), which uses numerical embeddings as features, this new version uses words as features, resulting in improved interpretability and performance, especially when few positive training instances are available. Txt2onto 2.0 uses embeddings from a large language model during prediction to deal with unseen-yet-relevant words in the input text and to highlight biomedical concepts in the input text that are related to each disease and tissue term being predicted, thereby explaining the basis of every annotation. We demonstrate the generalizability of txt2onto 2.0 by accurately predicting disease annotations for studies from independent datasets, using proteomics and clinical trials as examples. Overall, our approach can annotate biomedical text regardless of experimental types or sources. Code, data, and trained models are available at https://github.com/krishnanlab/txt2onto2.0.
-
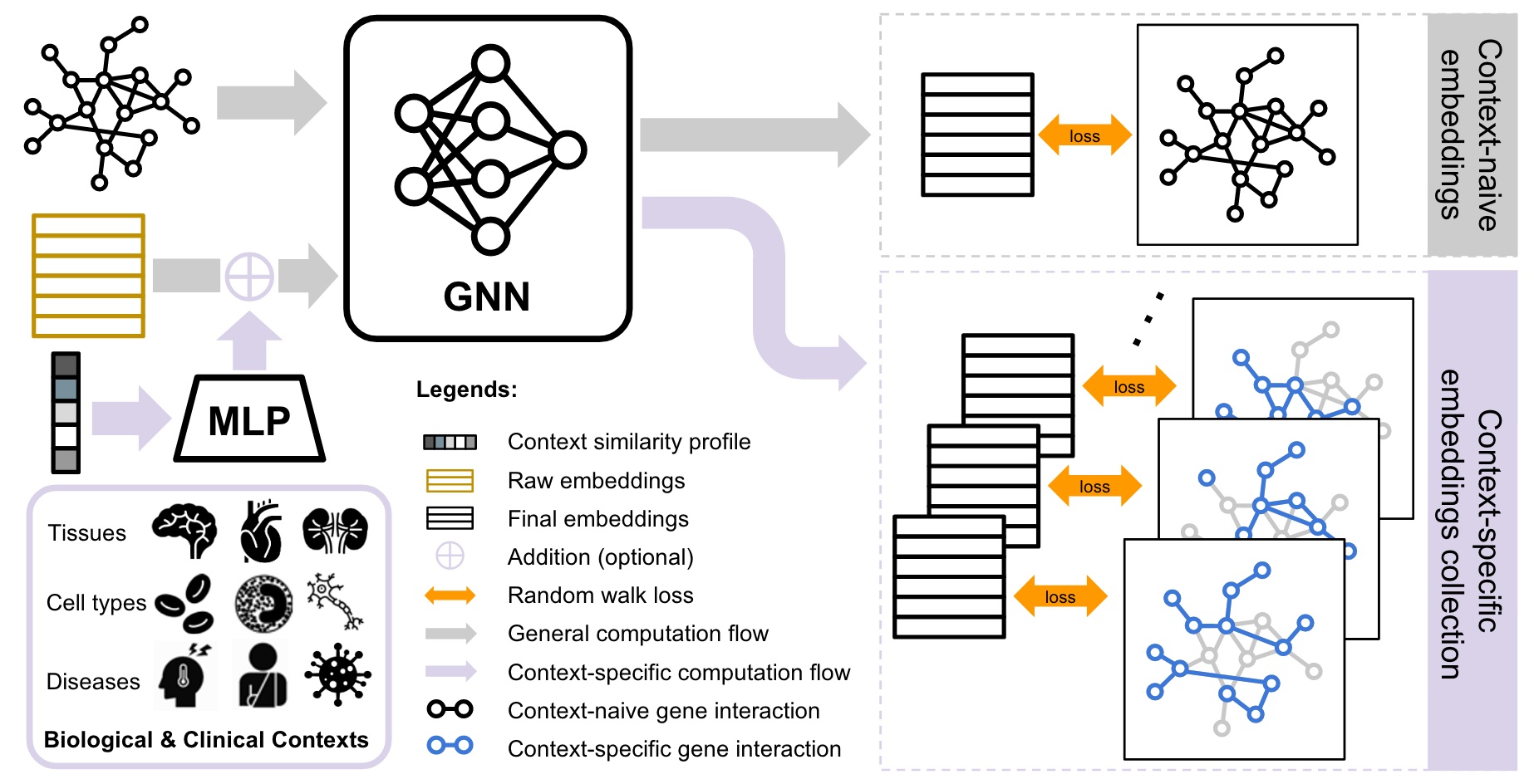 CONE: COntext-specific Network Embedding via Contextualized Graph AttentionRenming Liu, Hao Yuan, Kayla Johnson, and 1 more authorIn Proceedings of the 19th Machine Learning in Computational Biology meeting, 05–06 sep 2024
CONE: COntext-specific Network Embedding via Contextualized Graph AttentionRenming Liu, Hao Yuan, Kayla Johnson, and 1 more authorIn Proceedings of the 19th Machine Learning in Computational Biology meeting, 05–06 sep 2024Human gene interaction networks, commonly known as interactomes, encode genes’ functional relationships, which are invaluable knowledge for translational medical research and the mechanistic understanding of complex human diseases. Advanced network embedding techniques have inspired recent efforts to identify novel human disease-associated genes using canonical interactome embeddings. However, a pivotal challenge persists as many complex diseases manifest in specific biological contexts, such as tissues or cell types, while many existing interactomes do not encapsulate such information. Here, we propose CONE (\urlhttps://github.com/krishnanlab/cone), a versatile approach to generate context-specific embeddings from any context-free interactomes. The core component of CONE consists of a graph attention network with contextual conditioning, which is trained in a noise-contrastive fashion using contextualized interactome random walks localized around contextual genes. We demonstrate the strong performance of CONE embeddings in identifying disease-associated genes when using known associated biological contexts to the diseases. Furthermore, our approach offers new insights into the biological contexts associated with human diseases.



2022
-
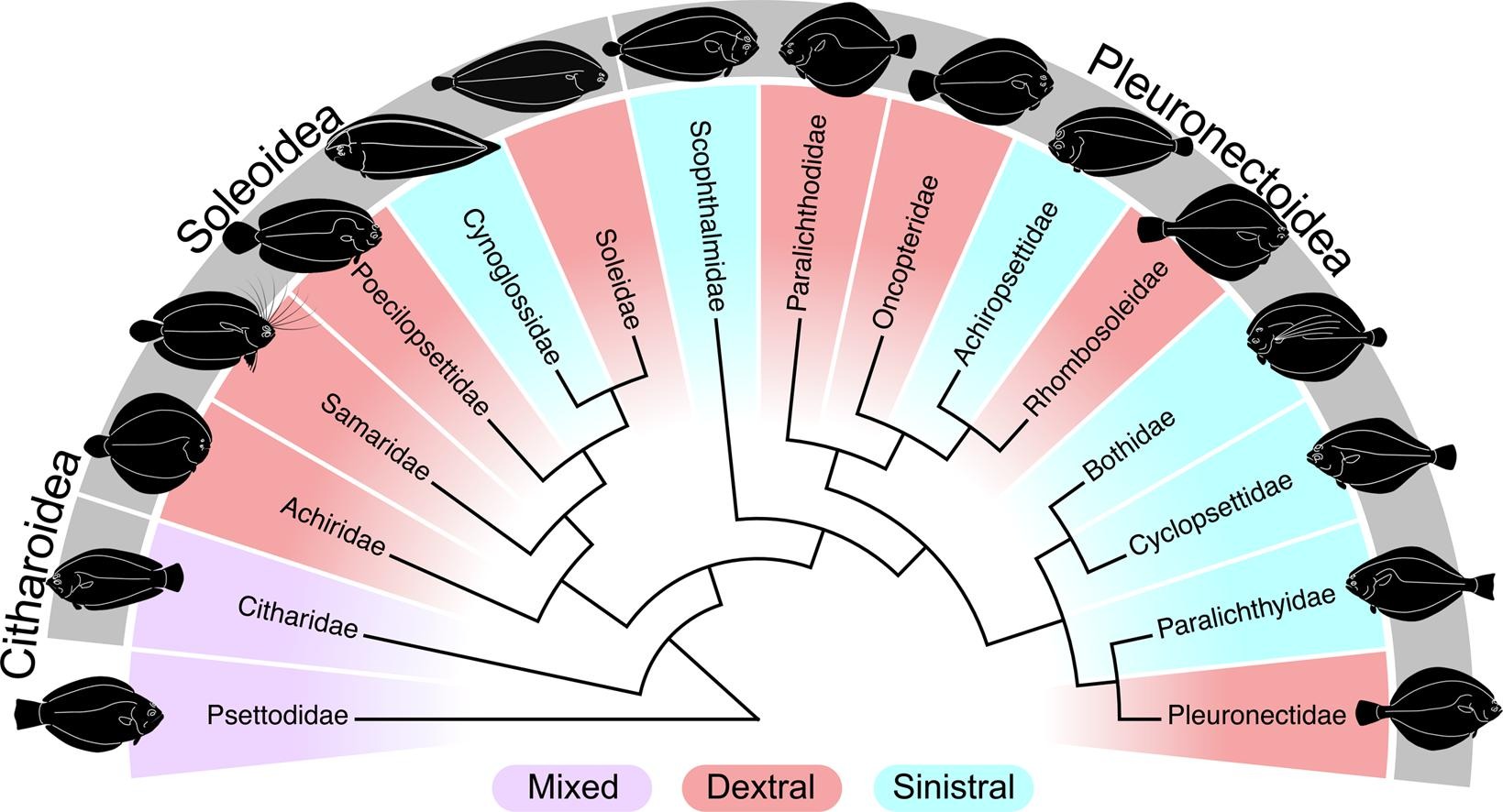 Exon-capture data and locus screening provide new insights into the phylogeny of flatfishes (Pleuronectoidei)Calder J. Atta, Hao Yuan, Chenhong Li, and 5 more authorsMolecular Phylogenetics and Evolution, Jan 2022
Exon-capture data and locus screening provide new insights into the phylogeny of flatfishes (Pleuronectoidei)Calder J. Atta, Hao Yuan, Chenhong Li, and 5 more authorsMolecular Phylogenetics and Evolution, Jan 2022There is an extensive collection of literature on the taxonomy and phylogenetics of flatfishes (Pleuronectiformes) that extends over two centuries, but consensus on many of their evolutionary relationships remains elusive. Phylogenetic uncertainty stems from highly divergent results derived from morphological and genetic characters, and between various molecular datasets. Deciphering relationships is complicated by rapid diversification early in the Pleuronectiformes tree and an abundance of studies that incompletely and inconsistently sample taxa and genetic markers. We present phylogenies based on a genome-wide dataset (4,434 nuclear markers via exoncapture) and wide taxon sampling (86 species spanning 12 of 16 families) of the largest flatfish suborder (Pleuronectoidei). Nine different subsets of the data and two tree construction approaches (eighteen phylogenies in total) are remarkably consistent with other recent molecular phylogenies, and show strong support for the monophyly of all families included except Pleuronectidae. Analyses resolved a novel phylogenetic hypothesis for the family Rhombosoleidae as being within the Pleuronectoidea rather than the Soleoidea, and failed to support the subfamily Hippoglossinae as a monophyletic group. Our results were corroborated with evidence from previous phylogenetic studies to outline regions of persistent phylogenetic uncertainty and identify groups in need of further phylogenetic inference.
-
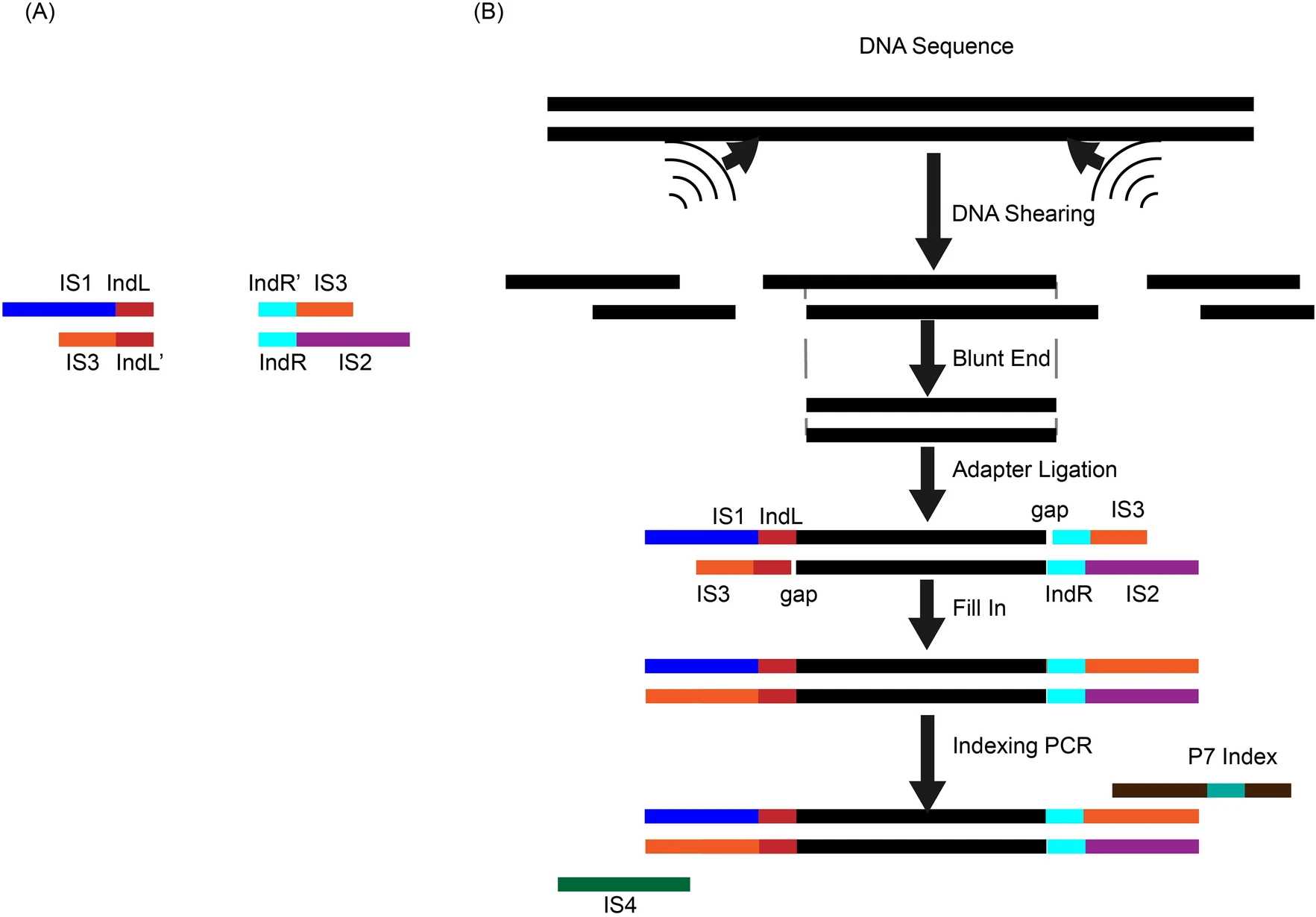 Inline index helped in cleaning up data contamination generated during library preparation and the subsequent stepsYing Wang, Hao Yuan, Junman Huang, and 1 more authorMolecular Biology Reports, Jan 2022
Inline index helped in cleaning up data contamination generated during library preparation and the subsequent stepsYing Wang, Hao Yuan, Junman Huang, and 1 more authorMolecular Biology Reports, Jan 2022Background High-throughput sequencing involves library preparation and amplification steps, which may induce contamination across samples or between samples and the environment. Methods We tested the effect of applying an inline-index strategy, in which DNA indices of 6 bp were added to both ends of the inserts at the ligation step of library prep for resolving the data contamination problem. Results Our results showed that the contamination ranged from 0.29 to 1.25% in one experiment and from 0.83 to 27.01% in the other. We also found that contamination could be environmental or from reagents besides cross-contamination between samples. Conclusions Inline-index method is a useful experimental design to clean up the data and address the contamination problem which has been plaguing high-throughput sequencing data in many applications.


2019
-
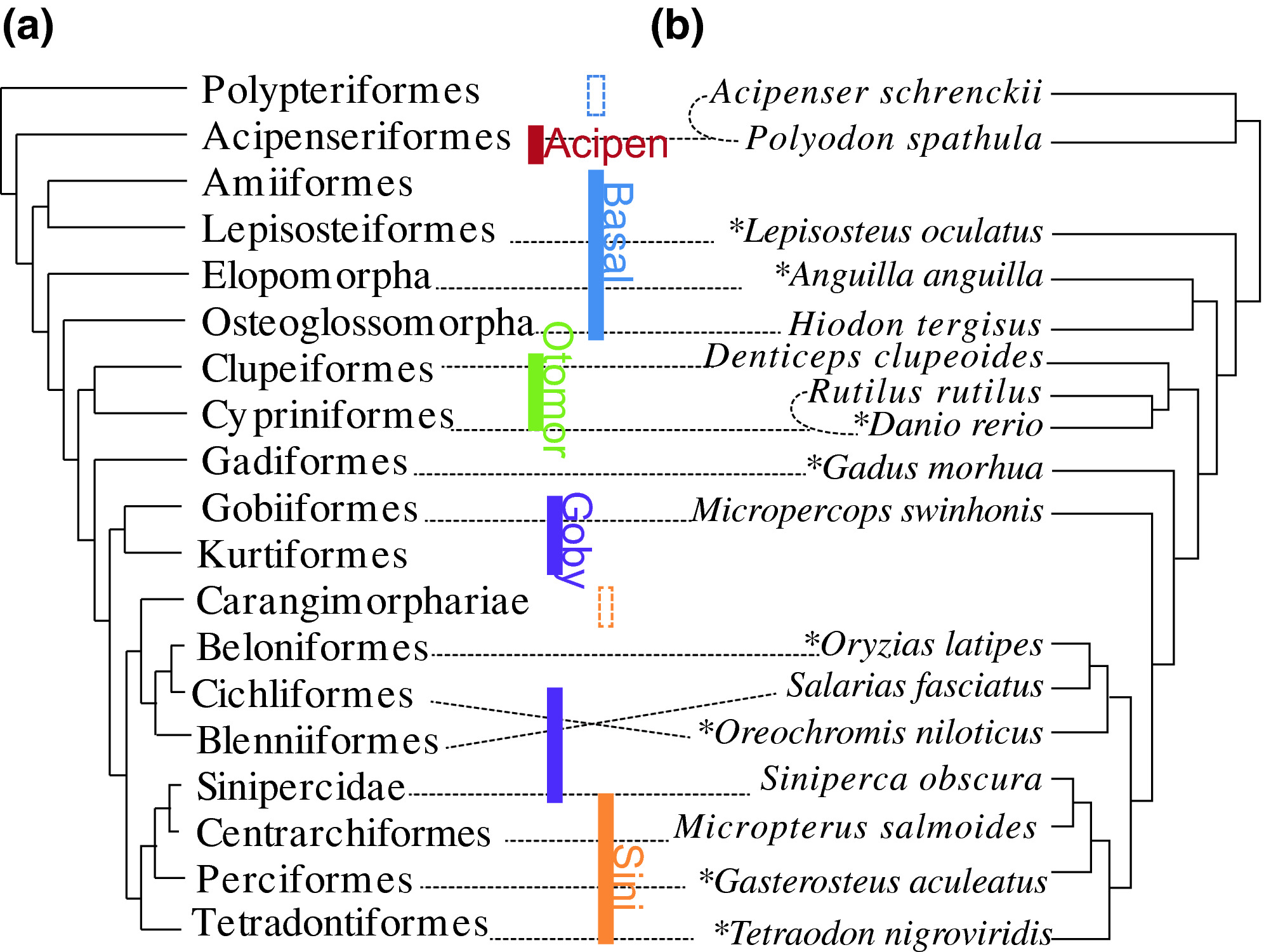 Gene markers for exon capture and phylogenomics in ray‐finned fishesJiamei Jiang, Hao Yuan, Xin Zheng, and 10 more authorsEcology and Evolution, Apr 2019
Gene markers for exon capture and phylogenomics in ray‐finned fishesJiamei Jiang, Hao Yuan, Xin Zheng, and 10 more authorsEcology and Evolution, Apr 2019Gene capture coupled with the next‐generation sequencing has become one of the preferred methods of subsampling genomes for phylogenomic studies. Many exon markers have been developed in plants, sharks, frogs, reptiles, fishes, and others, but no universal exon markers have been tested in ray‐finned fishes. Here, we identified a suite of “single‐copy” protein‐coding sequence (CDS) markers through comparing eight fish genomes, and tested them empirically in 83 species (33 families and nine orders or higher clades: Acipenseriformes, Lepisosteiformes, Elopomorpha, Osteoglossomorpha, Clupeiformes, Cypriniformes, Gobiaria, Carangaria, and Eupercaria; sensu Betancur et al. 2013). Sorting the markers according to their completeness and phylogenetic decisiveness in taxa tested resulted in a selection of 4,434 markers, which were proven to be useful in reconstructing phylogenies of the ray‐finned fishes at different taxonomic levels. We also proposed a strategy of refining baits (probes) design a posteriori based on empirical data. The markers that we have developed may greatly enrich the batteries of exon markers for phylogenomic study in ray‐finned fishes.
-
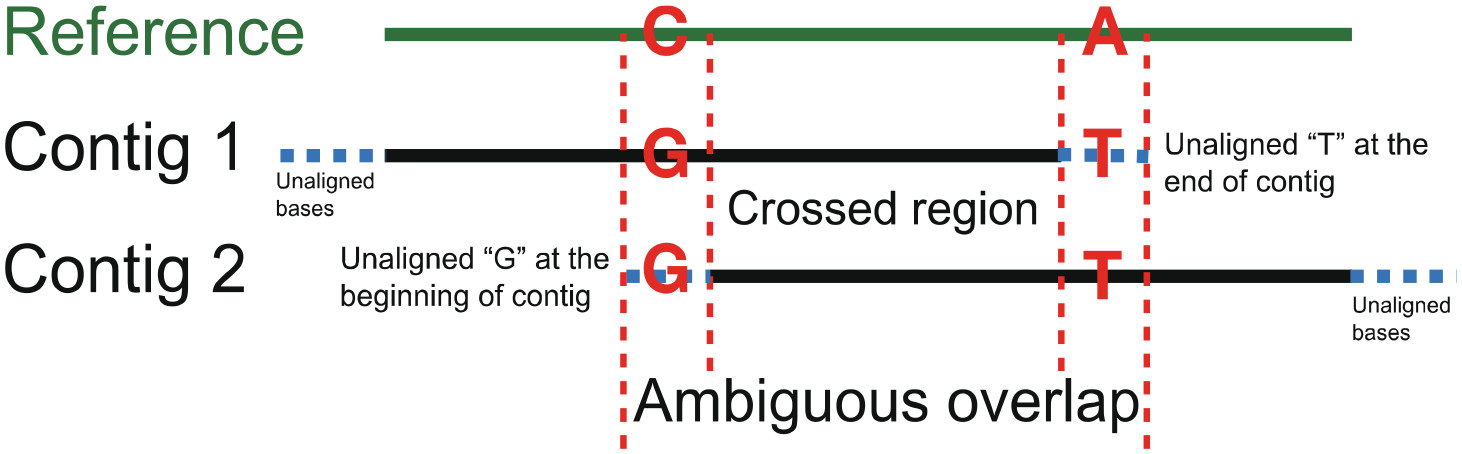 Assexon: Assembling Exon Using Gene Capture DataHao Yuan, Calder Atta, Luke Tornabene, and 1 more authorEvolutionary Bioinformatics, Jan 2019
Assexon: Assembling Exon Using Gene Capture DataHao Yuan, Calder Atta, Luke Tornabene, and 1 more authorEvolutionary Bioinformatics, Jan 2019Exon capture across species has been one of the most broadly applied approaches to acquire multi-locus data in phylogenomic studies of non-model organisms. Methods for assembling loci from short-read sequences (eg, Illumina platforms) that rely on mapping reads to a reference genome may not be suitable for studies comprising species across a wide phylogenetic spectrum; thus, de novo assembling methods are more generally applied. Current approaches for assembling targeted exons from short reads are not particularly optimized as they cannot (1) assemble loci with low read depth, (2) handle large files efficiently, and (3) reliably address issues with paralogs. Thus, we present Assexon: a streamlined pipeline that de novo assembles targeted exons and their flanking sequences from raw reads. We tested our method using reads from Lepisosteus osseus (4.37 Gb) and Boleophthalmus pectinirostris (2.43 Gb), which are captured using baits that were designed based on genome sequence of Lepisosteus oculatus and Oreochromis niloticus, respectively. We compared performance of Assexon to PHYLUCE and HybPiper, which are commonly used pipelines to assemble ultra-conserved element (UCE) and Hyb-seq data. A custom exon capture analysis pipeline (CP) developed by Yuan et al was compared as well. Assexon accurately assembled more than 3400 to 3800 (20%-28%) loci than PHYLUCE and more than 1900 to 2300 (8%-14%) loci than HybPiper across different levels of phylogenetic divergence. Assexon ran at least twice as fast as PHYLUCE and HybPiper. Number of loci assembled using CP was comparable with Assexon in both tests, while Assexon ran at least 7 times faster than CP. In addition, some steps of CP require the user’s interaction and are not fully automated, and this user time was not counted in our calculation. Both Assexon and CP retrieved no paralogs in the testing runs, but PHYLUCE and Hybpiper did. In conclusion, Assexon is a tool for accurate and efficient assembling of large read sets from exon capture experiments. Furthermore, Assexon includes scripts to filter poorly aligned coding regions and flanking regions, calculate summary statistics of loci, and select loci with reliable phylogenetic signal. Assexon is available at https://github.com/yhadevol/Assexon.


2018
-
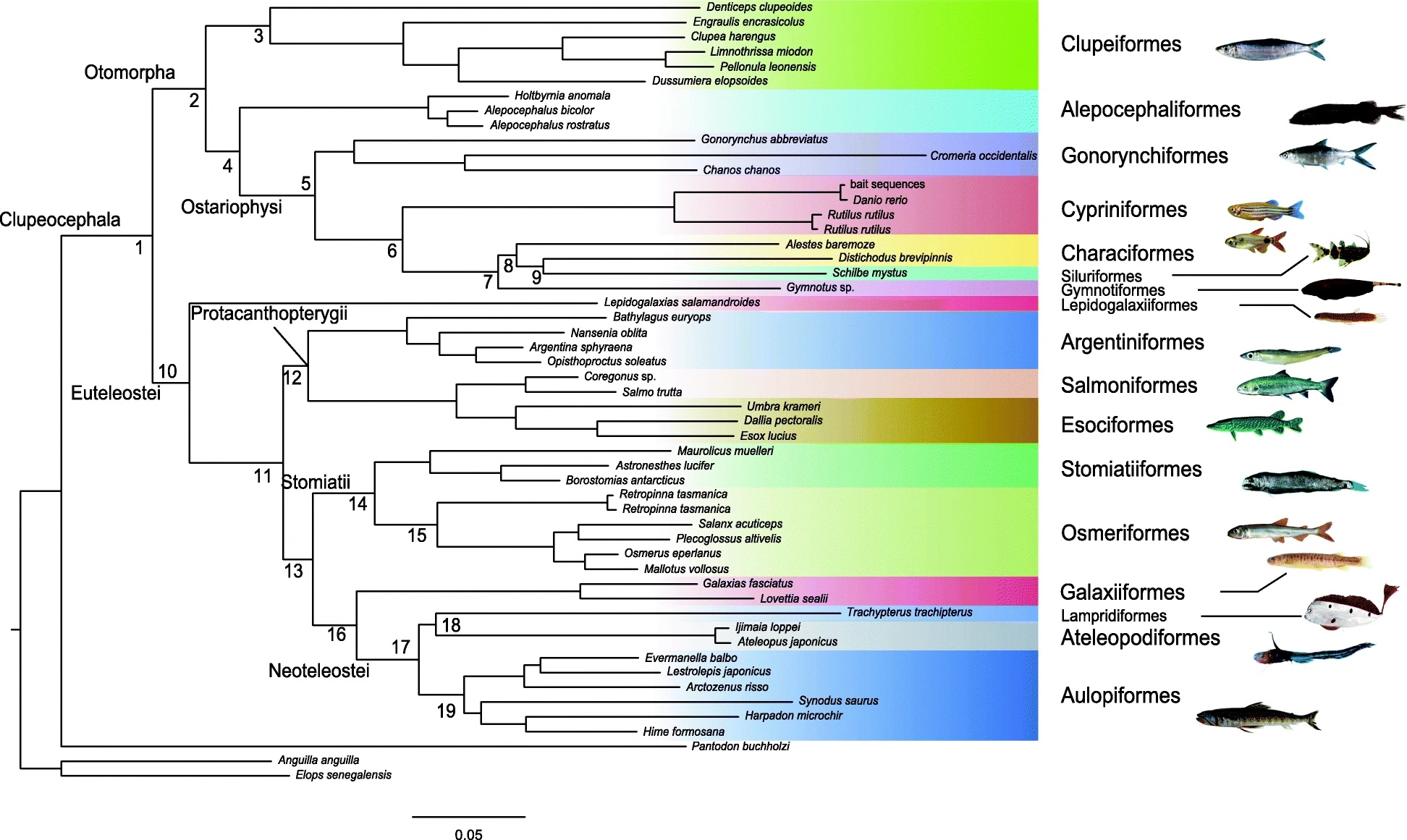 A phylogenomic approach to reconstruct interrelationships of main clupeocephalan lineages with a critical discussion of morphological apomorphiesNicolas Straube, Chenhong Li, Matthias Mertzen, and 2 more authorsBMC Evolutionary Biology, Dec 2018
A phylogenomic approach to reconstruct interrelationships of main clupeocephalan lineages with a critical discussion of morphological apomorphiesNicolas Straube, Chenhong Li, Matthias Mertzen, and 2 more authorsBMC Evolutionary Biology, Dec 2018Background: Previous molecular studies on the phylogeny and classification of clupeocephalan fishes revealed numerous new taxonomic entities. For re-analysing these taxa, we perform target gene capturing and subsequent next generation sequencing of putative ortholog exons of major clupeocephalan lineages. Sequence information for the RNA bait design was derived from publicly available genomes of bony fishes. Newly acquired sequence data comprising \textgreater 800 exon sequences was subsequently used for phylogenetic reconstructions. Results: Our results support monophyletic Otomorpha comprising Alepocephaliformes. Within Ostariophysi, Gonorynchiformes are sister to a clade comprising Cypriniformes, Characiformes, Siluriformes and Gymnotiformes, where the interrelationships of Characiformes, Siluriformes and Gymnotiformes remain enigmatic. Euteleosts comprise four major clades: Lepidogalaxiiformes, Protacanthopterygii, Stomiatii, and Galaxiiformes plus Neoteleostei. The monotypic Lepidogalaxiiformes form the sister-group to all remaining euteleosts. Protacanthopterygii, comprising Argentini-, Esoci- and Salmoniformes, is sister to Stomiatii (Osmeriformes and Stomiatiformes) and Galaxiiformes plus Neoteleostei. Conclusions: Several proposed monophyla defined by morphological apomorphies within the Clupeocephalan phylogeny are confirmed by the phylogenetic estimates presented herein. However, other morphologically described groups cannot be reconciled with molecular phylogenies. Thus, numerous morphological apomoprhies of supposed monophyla are called into question. The interpretation of suggested morphological synapomorphies of otomorph fishes is strongly affected by the inclusion of deep-sea inhabiting, and to that effect morphologically adapted Alepocephaliformes. Our revision of these potential synapomorphies, in the context that Alepocephaliformes are otomorph fishes, reveals that only a single character of the total nine characters proposed as synapomorphic for the group is clearly valid for all otomorphs. Three further characters remain possible apomorphies since their status remains unclear in the deep-sea adapted Alepocephaliformes showing developmental lag and lacking a swim bladder. Further, our analysis places Galaxiiformes as sister group to neoteleosts, which contradicts some previous molecular phylogenetic studies. This needs further investigation from a morphological perspective, as suggested synapomophies for several euteleostean lineages are challenged or still lacking. For the verification of results presented herein, a denser phylogenomic-level taxon sampling should be applied.

2016
-
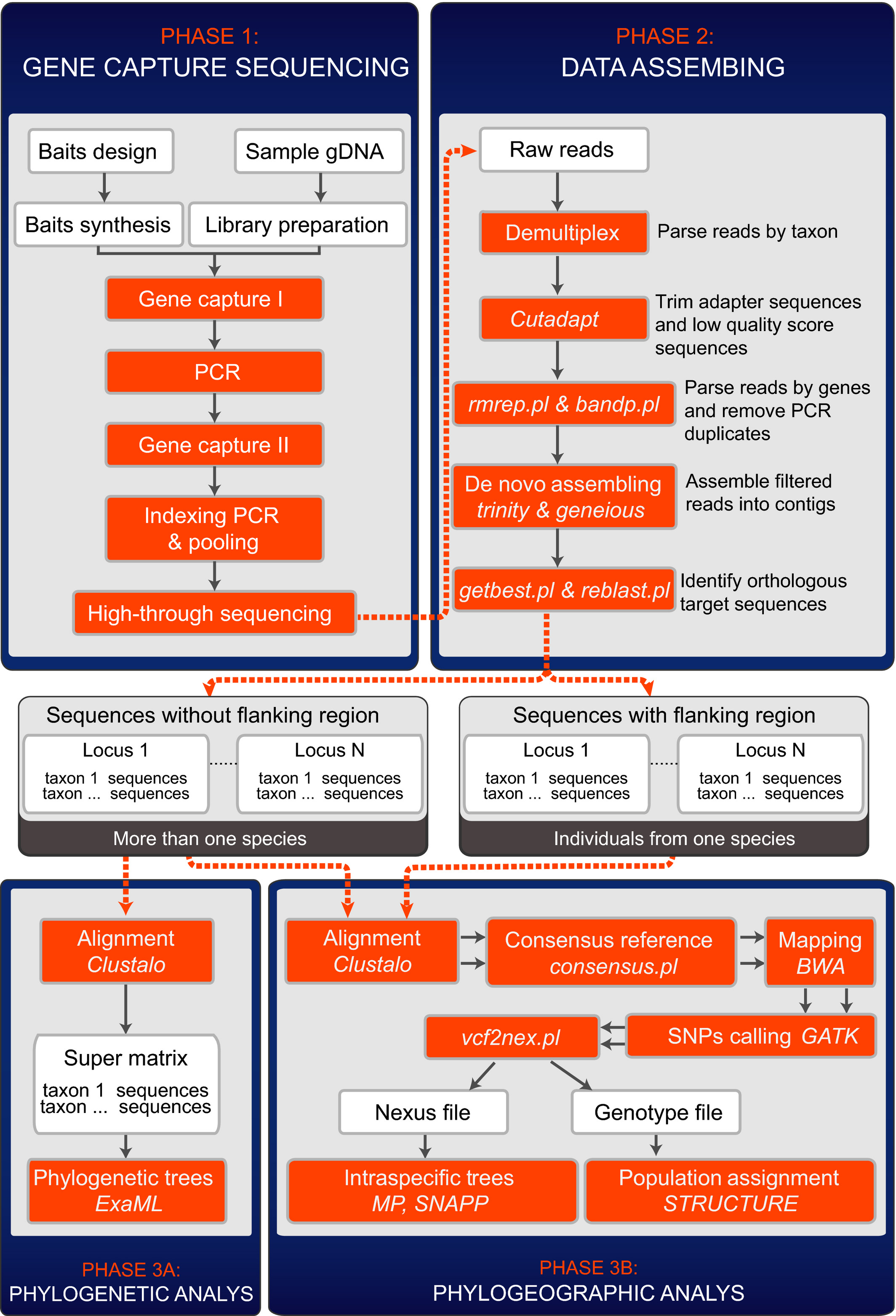 Target gene enrichment in the cyclophyllidean cestodes, the most diverse group of tapewormsHao Yuan, Jiamei Jiang, Francisco Agustín Jiménez, and 4 more authorsMolecular Ecology Resources, Sep 2016
Target gene enrichment in the cyclophyllidean cestodes, the most diverse group of tapewormsHao Yuan, Jiamei Jiang, Francisco Agustín Jiménez, and 4 more authorsMolecular Ecology Resources, Sep 2016The Cyclophyllidea is the most diverse order of tapeworms, encompassing species that infect all classes of terrestrial tetrapods including humans and domesticated animals. Available phylogenetic reconstructions based either on morphology or molecular data lack the resolution to allow scientists to either propose a solid taxonomy or infer evolutionary associations. Molecular markers available for the Cyclophyllidea mostly include ribosomal DNA and mitochondrial loci. In this study, we identified 3641 single-copy nuclear coding loci by comparing the genomes of Hymenolepis microstoma, Echinococcus granulosus and Taenia solium. We designed RNA baits based on the sequence of H. microstoma, and applied target enrichment and Illumina sequencing to test the utility of those baits to recover loci useful for phylogenetic analyses. We captured DNA from five species of tapeworms representing two families of cyclophyllideans. We obtained an average of 3284 (90%) of the targets from the test samples and then used captured sequences (2 181 361 bp in total; fragment size ranging from 301 to 6969 bp) to reconstruct a phylogeny for the five test species plus the three species for which genomic data are available. The results were consistent with the current consensus regarding cyclophyllidean relationships. To assess the potential for our method to yield informative genetic variation at intraspecific scales, we extracted 14 074 single nucleotide polymorphisms (SNPs) from alignments of four Arostrilepis macrocirrosa and two A. cooki and successfully inferred their relationships. The results showed that our target gene tools yield data sets that provide robust inferences at a range of taxonomic scales in the Cyclophyllidea.
